![[exit]](/exit-outline.svg "Leave")
This post will discuss the potential of HDAC6 inhibition in relevance to cognitive enhancement and reduction of neurodegenerative disorders.
HDAC6 
Histone deacetylases, commonly known as HDACs, are a class of enzymes that hold relevance to cognitive and physiological factors in the body. They operate by removing acetyl groups from histones and other protein regulatory factors, with functional consequences on chromatin remodeling and gene expression profiles. [1]
There are 11 HDAC subtypes, ranging from HDAC1-11. There are many unselective HDAC inhibitors, and they have some benefits, but the unselectivity for HDAC subtypes causes many side effects which limits the tolerable dose ceiling [4] and also efficacy especially in cognitive enhancement. To attenuate this issue, aiming for higher selectivity is key, and that is why targeting the 6th HDAC subunit is interesting.
The reason for HDAC6 specifically over other types is because of its interactions with microtubule dynamics, but it is quite complex so we will get onto that later. Concerning other HDAC subunits, HDAC -2 and -3 are seemingly relatively attractive to inhibit as well as HDAC6, and may serve in a useful combination if targeted at the same time.
To start with basal location, HDAC6 is mainly located in the Cytoplasm. [1]
It has been also reported recently that HDAC6 may have a role in neuronal development and function . Studies of HDAC inhibitors (HDACi) in human neuronal cells show that HDAC6 inhibitors (HDAC6i) increase the acetylation of specific lysine residues in proteins involved in synaptogenesis. [2]
In addition to the role of HDAC6 in neurons, animal studies show a role for HDAC6 in Oligodendrocytes. Cultured rat Oligodendrocytes were shown to express HDAC6 and inhibition of HDAC6 by Tubastatin A resulted in decreased microtubule binding activity of tau. It was shown that HDAC6 inhibition led to increased acetylation of tau in the Oligodendrocytes, which in turn reduced its turnover rate [2]
HDAC6 inhibition also interacts with the dopamine and limbic circuits. In one study, it ameliorated behavioral impairment caused by Meth, improving locomotor deficit and reducing DA and DOPAC depletion. HDAC6 inhibition effectively suppressed the death of dopaminergic neurons [10]
Another interesting note about the neuroprotective properties of HDAC6 inhibition is its ability to inhibit α-syn accumulation [10]. The excessive accumulation of insoluble α-syn, which is toxic to dopaminergic neurons, is considered an important event in the pathogenesis of PD.
For other effects, HDAC6 inhibition has also been found to promote antidepressant-like activity in vivo [11], improving models associated with depression and anxiety.

HDAC6 inhibition is beneficial for many neurodegenerative disorders, but it seems most effacious or most proven for ameleorating Alzheimer's disease. One issue though, is that it may be bidirectional in effects. While acutely it is nearly always neuroprotective, in tauopathy models it can actually be inhibitory.
A HDAC6-dependent surveillance mechanism suppresses toxic tau accumulation, which may protect against the progression of AD and related tauopathies. [12]
For example, acute pharmacological HDAC6 inhibition with tubastatin A (TBST) alleviated behavioral and cognitive deficits in transgenic tau or Aβ mouse models, spurring significant interest in using HDAC6 inhibitors as AD therapies. However, we previously showed that acetylated tau is a substrate for HDAC6 and that inhibition of HDAC6 could result in elevated levels of acetylated tau. Thus, chronic long-term HDAC6 inhibition or depletion could conceivably increase acetylated tau, promote tau aggregation, and lead to cognitive defects. [12]
Also, HDAC6 can stabilize microtubules by deacetylating α-tubulin, and can control p53 activity by targeting acetyl-Lysine 381/382. α-tubulin is defined as a protein that interacts with beta tubulin to form heterodimers, which are essential components of microtubules.
Our evidence reveals that HDAC6 localizes to the perinuclear and leading-edge subcellular regions and is associated, in part, with microtubules. The co-localization with the p150glued-containing motor complex indicates that HDAC6 might control microtubule motor-based cargo transport or sorting by modulating the acetyl- ation of specific proteins. [3]
HDAC6 is also important for α-tubulin lactylation, with HDAC6 overexpression leading to increased α-tubulin lactylation, and HDAC6 deficiency causing a significant reduction in α-tubulin lactylation [13].
With the information available, HDAC6 inhibition might be most useful in short, acute cycles, similar to the approach of ICOT [14], rather than chronic use. It may also best be combined with approaches that reduce tauopathy to reduce potential downsides.
Inhibitor Candidates
There are many HDAC inhibitors, but most of them are not selective. The most efficacious "unselective" ones actually usually are, but to around 2-3 HDAC subtypes, such as for HDAC2 & HDAC3. There are still issues with the unselectivity of current HDAC inhibitors [4] (such as tolerability) so improved candidates should be focused on.
The following are the candidates for HDAC6 inhibition based on current research:
To start with SW-100, it seems to be a good candidate. It has mostly ideal pharmacokinetics, and high selectivity and potency with an IC50 of 2.3nM [6]. However, it has one main issue, which is a short half life [7]. For this reason it makes it unoptimal and it has caused issues in studies where it has been used as the effects don't last a full day.
Tubastatin A has similar issues with a half life of 2 hours, making it not very viable [11].
CAY10603 is another interesting molecule. It is a relatively large molecule [8] however it makes up for it by being extremely potent with an IC50 of 2pM [9] (~1000x the affinity of SW-100). It also has high selectivity over other HDACs.
It has a similar problem as SW-100 however, which is a short half-life. A solution for this was found in one study which designed PLGA CAY10603-loaded nanoparticles, with the modification known as PLGA@CAY@Lf NPs [10]. Apart from just extending half life, the modification to base CAY (CAY10603) showed a significantly greater effect in increasing acetyl-α-tubulin than PLGA@CAY NPs (1.3-fold, p < 0.05), meaning it likely has improved efficacy as well.
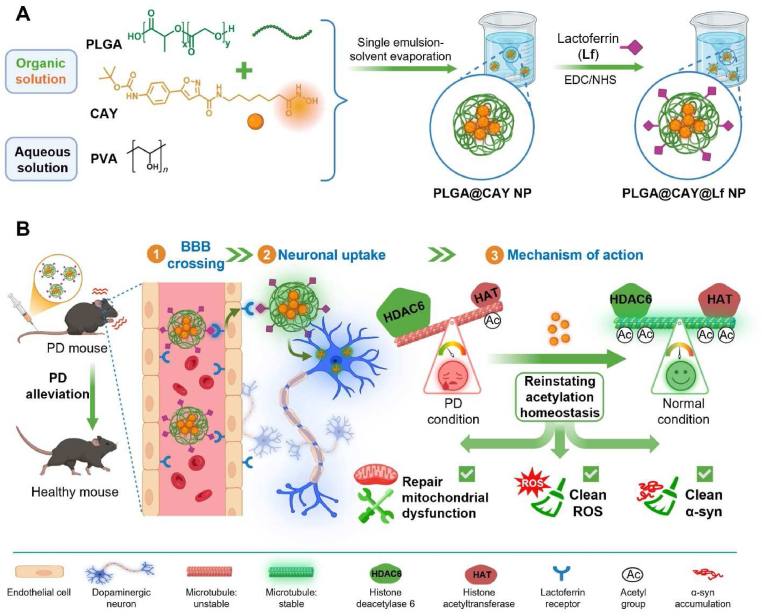
LGA@CAY@Lf NPs demonstrated enhanced BBB penetration and significant brain accumulation by leveraging on the brain targeting properties of Lf. Importantly, CAY released from PLGA@CAY@Lf NPs restored the disrupted acetylation balance in PD, resulting in neuroprotection through the reversal of mitochondrial dysfunction, ROS elimination, and inhibition of α-syn accumulation. Furthermore, PLGA@CAY@Lf NPs treatment recovered TH and DA levels to near-normal levels, alleviated neuroinflammation, and markedly improved behavioral impairments. [10]
Review
With information available, LGA@CAY10603@Lf NPs may be the best approach for HDAC6 inhibition because of its ideal pharmacokinetics. The modification could be dosed not often, such as once a month, as it may be optimal to reduce chronic administration with the bidirectional dynamics of HDAC6 inhibition.
Acutely HDAC6i is neuroprotective but strangely potentially degenerative in conditions of high tau - HDAC6i isn't really the problem, aberrant tau is. The pathway seems to require multiple other combined approaches to reduce potential downsides while maximizing upsides of HDAC6i.
HDAC6 should definitely be the focus of more research for how to maximize use of the pathway without negatives.
Thanks for reading, contributions are welcome.