![[exit]](/exit-outline.svg "Leave")
Introduction 
The Dorsolateral Pre-Frontal Cortex (dlPFC) is one of the most interesting parts of the brain. It has very significant implications on spatial cognition and working memory, visualizing, executive function and more. It is one of the most important, yet one of the most underfocused areas for the goal of increasing cognitive potential.
Rodents do not have rostral PFC areas (e.g. Frontal Pole) or a dlPFC, so it makes it more difficult to study due to the requirement for primates or humans to study upon. Due to these reasons, and the focus for cognition being mainly on the hippocampus and other areas of the brain, the dlPFC and nearby regions seem more underresearched than they should be.
The dlPFC is one of the most recently evolved/developed areas of the brain, in close quarters with the Frontal Pole Cortex (FPC/Brodmann area 10/10p). The dlPFC is much more dependent (~5x more dependent) on NMDA-NR2b than AMPA compared to other parts of the cortex, in this case it is compared against the Visual Cortex (for Delay Cell Firing).
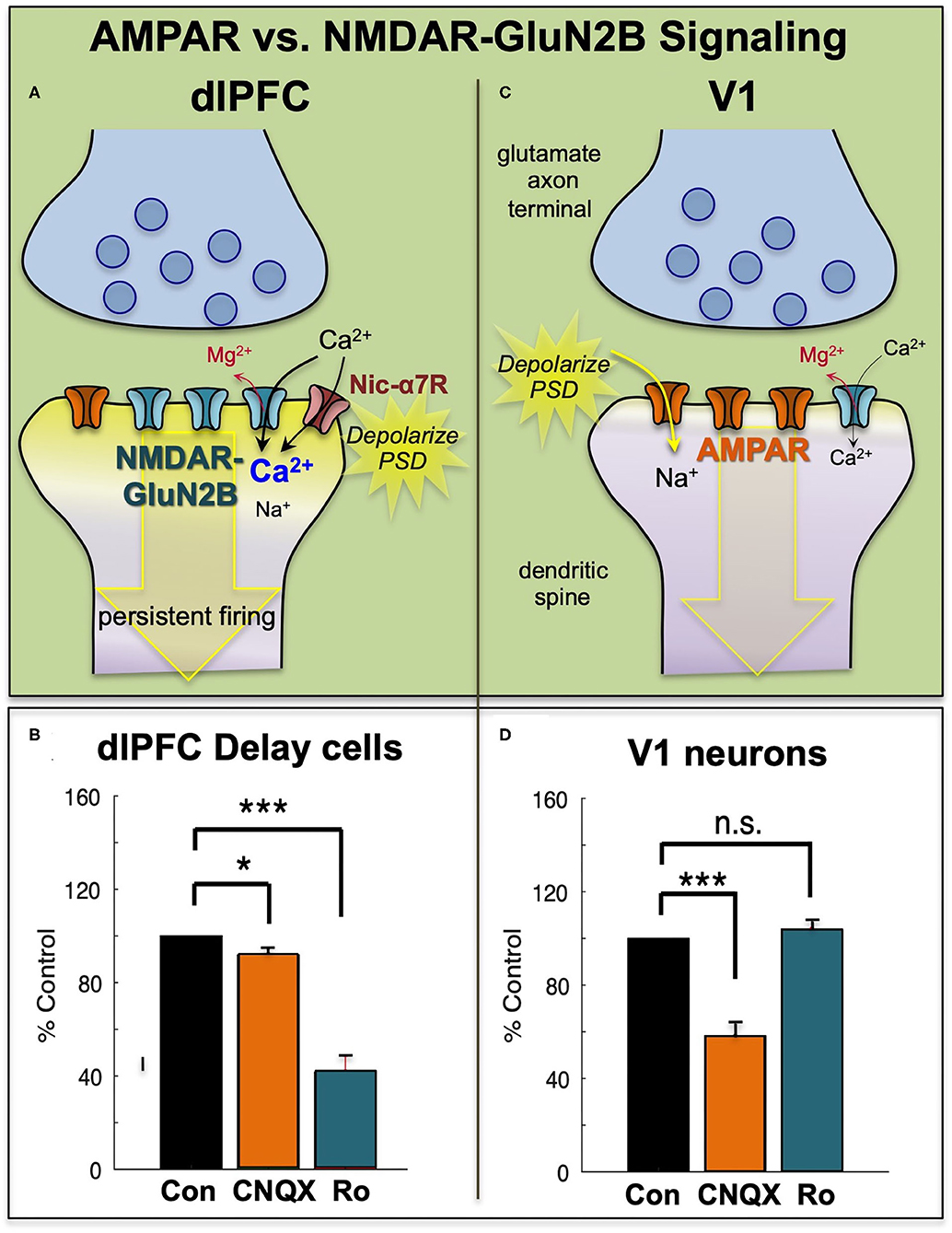
This is part of the reason why the dlPFC and surrounding areas are so vulnerable to aging-based deteriation. -tbf-
The dlPFC has a lot of important functions, but something to note as we start, is that the dlPFC is heavily impacted by mGluR2 and mGluR3 firing. Modulating either of these, directly, or via other mechanisms like NAAG or GCP-II, has significant impacts on Delay Cell Firing and the function of the dlPFC (working memory, cognition, etc).
Delay Cells (which will be mentioned often) reside in pyramidal cell microcircuits in deep layer III (the most cognitively important layer) of the dlPFC that have extensive recurrent excitatory connections.
Table of Contents
dlPFC Neuroscience:
- About the dlPFC
- Relevance of Delay Cells
The Top-Down Control Dichotomy
- The relevance of the dlPFC for overlooking other parts of cognition
- Emotion and pain regulation
- Potential of dlPFC modulation for top-down control
mGluR Modulation:
- On NAAG/GCP-II and relevance
- Observed effects of mGluR3 modulation
- Cognition and fine tuning glutamate
- Protection against age-based decline
- Ameriolation for those with disease
Functions of the dlPFC
The primate dlPFC has the remarkable ability to generate and sustain mental representations without sensory stimulation, the foundation of abstract thought.
dlPFC “Delay cells” are able to maintain persistent firing across the delay period in a working memory task, sustaining representations over many seconds e.g., remembering a position in visual space [3]. The persistent firing of Delay cells across the delay period depends on NMDAR stimulation [4], a finding predicted by computational models (33). Thus, iontophoresis (local electrical application) of low doses of NMDAR antagonists, including antagonists that selectively block those with GluN2A or GluN2B subunits, markedly reduces Delay cell firing.
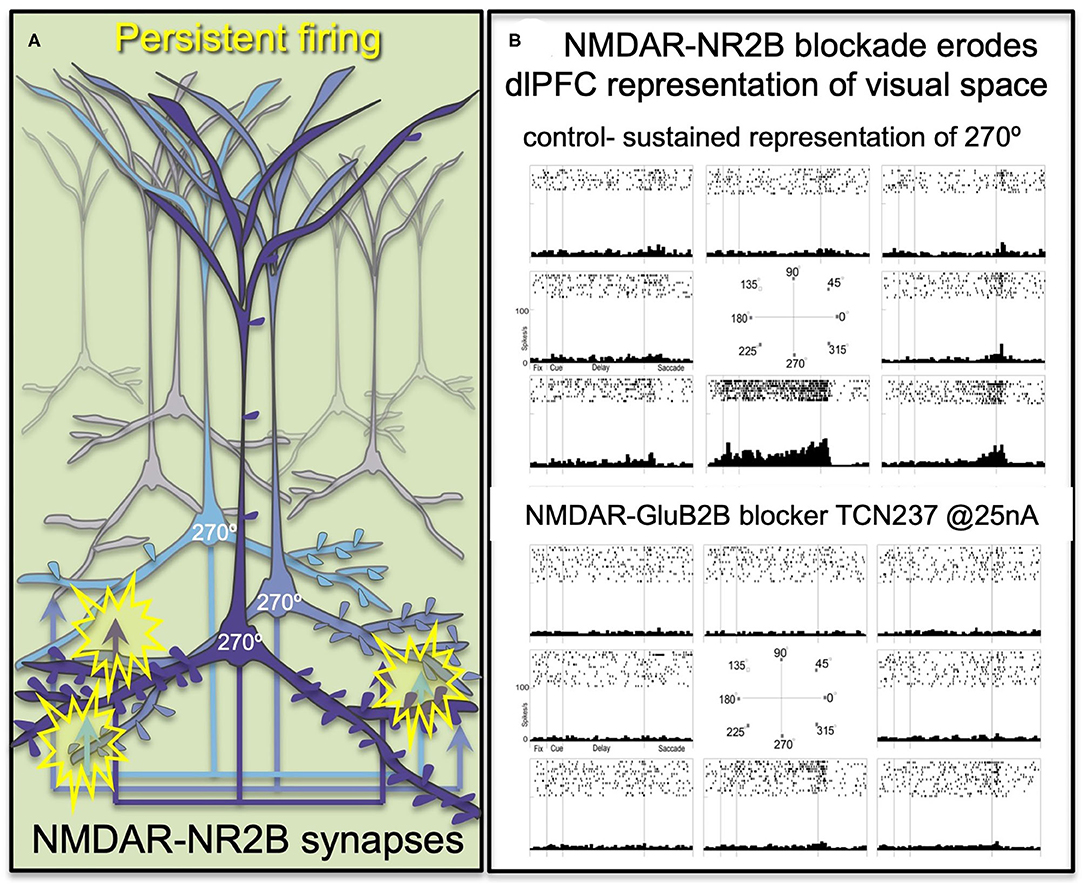
"The selective NMDAR- GluN2B antagonist, TCN237, completely blocks the ability of the neuron to generate representations of visual space."
In another study, dlPFC damage was associated with deficits in the manipulation of verbal and spatial knowledge, with left dlPFC necessary for manipulating information in working memory and right dlPFC critical for manipulating information in a broader range of reasoning contexts. [Source]
The dlPFC has functions that effect OCD. In a few studies [o1][o2][o3] modulating the left dlPFC using rTMS (Transcranial magnetic stimulation), they showed improvements in symptoms for OCD. However, positives have yet to be replicated using GCP-II inhibition or compound-based modulation.
The dlPFC also has implications in detecting threats [Source], pattern recognition, consciousness/consciousness awareness [Source], risk taking, mental health (depression, anhedonia, ADHD, schizo), possibly even physical endurance due to effort-cost judgements [Source] and many other things. So, lets look into it.
The Top-Down Control Dichotomy
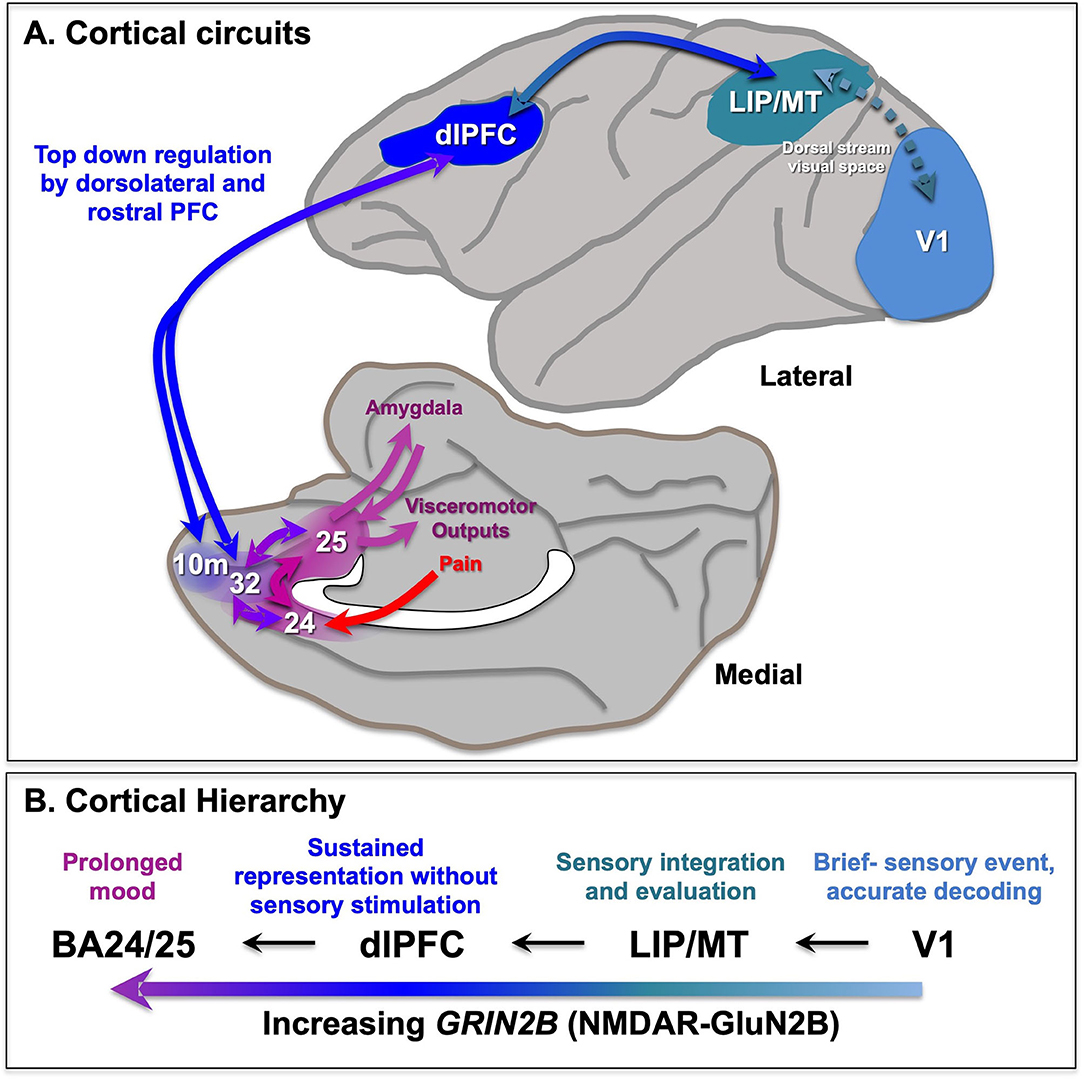
The dlPFC interacts through BA10m (close to the PFC) and BA32 to eventually communicate with the amagdyla. The more newly evolved, rostral and lateral areas of PFC provide top-down regulation of the more primitive medial and caudal areas. The dlPFC is a key structure for executive and attentional control whereby any transient (stressors, neurostimulation) or permanent (lesion) impairment compromises adaptive behavior [Source].
For example, the dlPFC can regulate emotion via direct projections to BA24 [1][2], and indirect projections to BA25 via BA10m or BA32 to BA25.
The dlPFC provides top-down regulation of emotion through indirect projections to BA25 via areas BA10m and BA32, and direct projections to BA24 (not shown) [Source].
Under conditions of stress or depression, elevated activity in the cingulate cortices can activate the amygdala, and very high levels of catecholamine release in cortex takes higher PFC areas such as dlPFC “offline.” The effects of dopamine hyperactivation and hypersexual stimuli likely work via this same pathway too effect too. Thus, there is a self-perpetuating, unregulated state, where primitive circuits prevail.
It seems easier for women to lose this top-down control compared to men. In women, the interaction between emotion and working memory yielded a significantly stronger response in the amygdala and the orbitofrontal cortex (OFC) compared to their male counterparts. [Source]
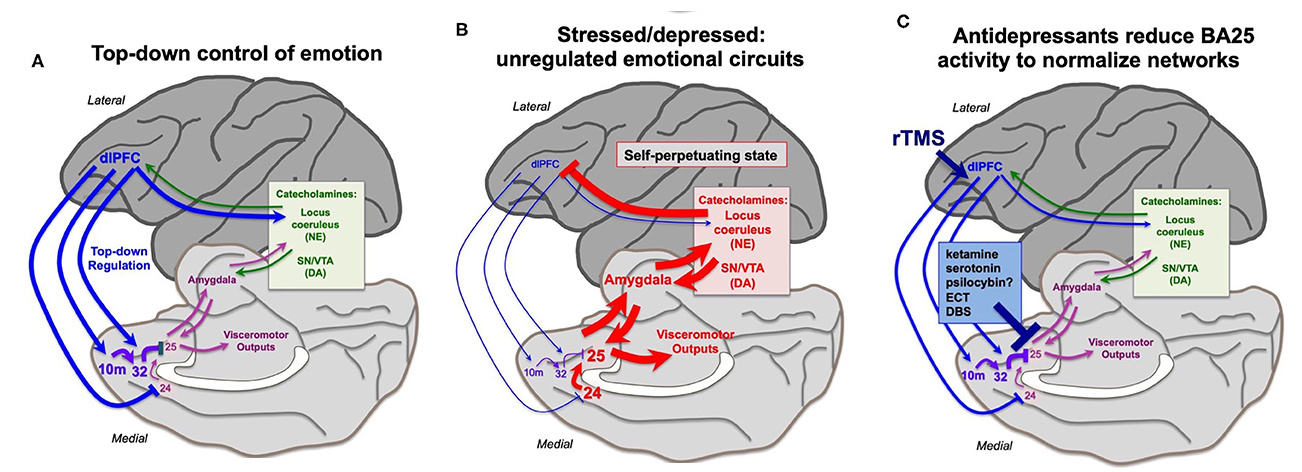
"Many antidepressant treatments (seretonin modulators, psychedelics, maybe even pinealon) reduce the activity of BA25. This may give the cortex a “foot in the door” to restore top-down regulation, especially when treatments promote dendritic spine restoration in higher PFC circuits. Other treatments may directly enhance the top-down regulation by the left dlPFC, e.g., rTMS and insight therapies."
The anti-addictive, anti-hedonistic, and pro-executive function of optimal dlPFC firing has been shown consistently across many studies. Here are some examples:
-
"Excitatory neuromodulation targeting the dorsolateral prefrontal cortex appears to lead to a sustained reduction of craving and consumption in individuals with addiction or overeating behaviour." [Source]
-
"The group which received active DLPFC tDCS showed significantly improved task performance across all EFs immediately after and 1 month following the intervention, when compared to both pre-stimulation baseline and individuals who received sham tDCS. Similarly, a significant reduction in craving was observed immediately after and 1 month following the intervention in the active, but not sham stimulation group." [Source]
-
"Pretreatment with 2-PMPA only partially attenuated cocaine-enhanced extracellular NAc DA but completely blocked cocaine-enhanced extracellular NAc glutamate, suggesting an important role of NAc glutamate in mediating the pharmacological action of 2-PMPA." [Source]
It is ever-more seeming like the dlPFC-amygdala connection is the connection containing the "gatekeeper" of the brain. We can have good intentions within the Cortex/dlPFC/surrounding areas, such as to start new habits or to do things that bring greater meaning and fulfilment. However, the Basal Ganglia/Amygdala can block these actions due to emotion/pain-based or effort-based means.
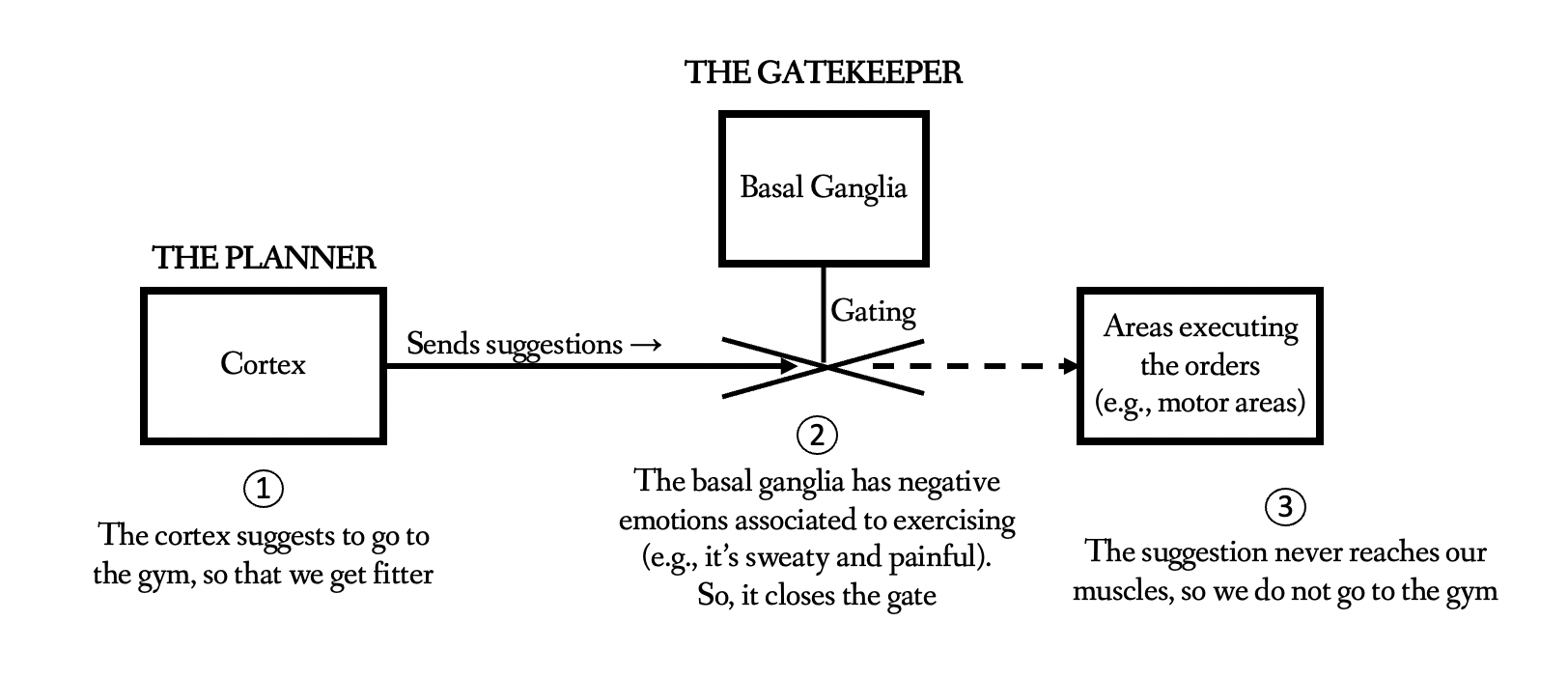
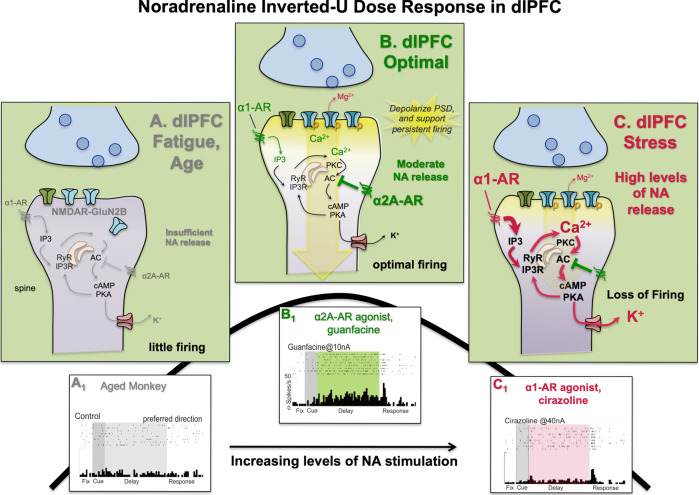
Guanfacine (which increases dlPFC delay cell firing through α2A-adrenergic agonism) has already been shown to change the dlPFC-amgydala relationship to enhance cognitive control [Source], however its upside has been limited by side effects of α2A-adrenergic receptor agonism, though it is still useful.
Amygdala overactivation lowers cognitive control and therefore is imparing to high level productivity and personal fulfilment. If we can keep top-down control, without letting primitive networks decide what we do based on immediate pleasure, then our higher intelligence can be more in control. This is incredibly desirable, from a neurological sense, to a philosophical sense. Being able to act intelligently without reverting to primitive pleasures, hedonism, and addiction, would amount to an evolution in the human form, especially if done through a compound modulating the dlPFC to a level of high significance.
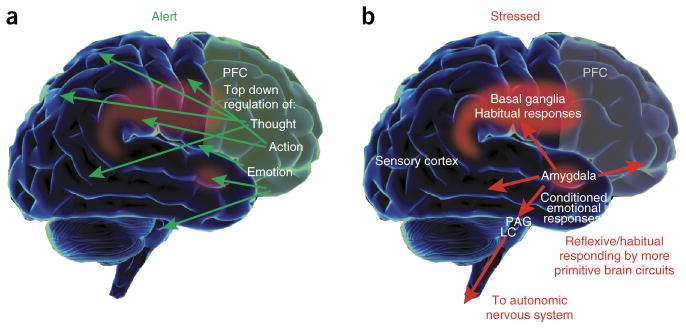
The dlPFC is a major component of motivation/anticipation and goals. The dorsolateral PFC (dlPFC) integrates and transmits signals of reward to the mesolimbic and meso-cortical DA circuits and initiates motivated behavior [Source].
This then shows the importance of the dlPFC in regulating emotion and top-down control/executive function. However, just BA25 modulation alone has its limits. Lowering BA25 activity may lower the control of the amagyla over the higher cortex areas, but how can we truly maximize the function of the dlPFC so we can have the highest amount of cognitive control and Delay Cell Firing?
We can do this by modulating mGluR2/3 (also known as group II mGluRs) interactions, which seem to have the largest effect on the function of the dlPFC.
Group II mGluRs (mGlu2/mGlu3)
mGluR2 interacts with mGluR3 to control Delay Cell Firing in the dlPFC. Group II mGluRs are primarily presynaptic autoreceptor with the function to inhibit glutamate transmission. However, both mGluR2 and mGluR3 can be found postsynaptically in regions such as dlPFC and Golgi cells respectively. mGluR3 can even be localized glially with astrocytic processes. In DLPFC, while mGluR2 is mostly located presynaptically as expected mGluR3 is mostly located here postsynaptically.
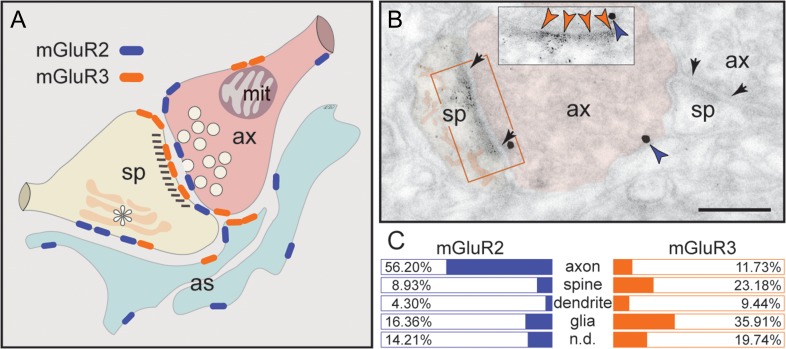
Cognitive deficits in disorders such as schizophrenia, aging, and Alzheimer's are increasingly associated with insults to mGluR3 receptors, while reductions in mGluR2 appear protective. This has been perplexing, as mGluR3 has been considered glial receptors, and mGluR2 and mGluR3 have been thought to have similar functions, reducing glutamate transmission. However, all the evidence points to mGluR2 and mGluR3 having opposing functions.
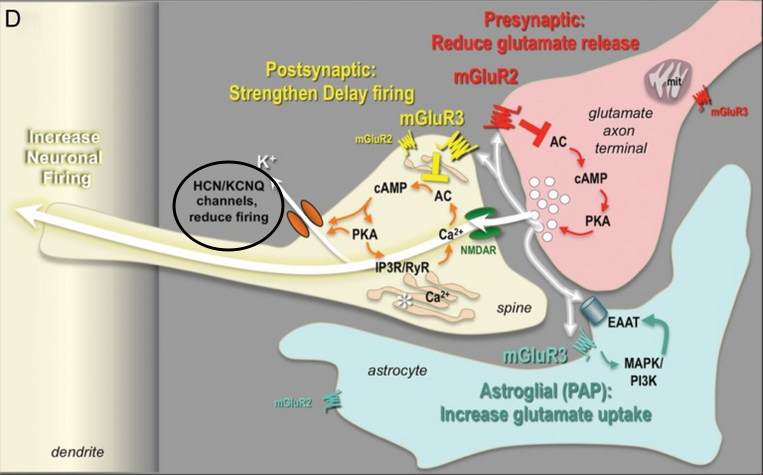
mGluR2 reduces Delay Cell Firing and glutamate, while mGluR3 increases Delay Cell Firing. Therefore, by increasing mGlu3 activity and lowering mGlu2 activity, we can increase Delay Cell Firing, which in turn increases the function of the dlPFC.
Something to note is that there are some interesting candidates for dual mGluR3 PAM / mGluR2 NAM actions. These dual mechanism compounds are very novel, with a lack of studies and they do not have the greatest safety/compound property predictions either. One candidate is known as PHCCC(4Me), also known as "THCCC" (not to be confused with PHCCC), however its safety predictions are not great. The best looking compound for dual mGluR3 PAM and mGluR2 NAM seems to be N-(4-methylphenyl)-7-oxo-1,7a-dihydrocyclopropa[b]chromene-1a-carboxamide, however it is unlikely to have any literature anytime soon. More information on these dual MOA compounds can be found here.
Increasing mGluR3 firing, while simultaneously reducing mGluR2 firing seems more beneficial than individual modulation of either. They seem to balance out potential issues of each other very well.
mGluR3 Modulation
Updated writeup:
This is a good time to talk about NAAG (N-Acetylaspartylglutamic Acid) and GCP-II (Glutamate Carboxypeptidase II/NAAG peptidase).
NAAG is an endogenous neurotransmitter that increases mGluR3 activity through agonism. NAAG is thought to be agonist of mGluR3 and with some actions on NMDA however it is debated its due to contamination with glutamate as purified NAAG had non-agonistic activity in one study suggesting it might be a PAM, while other study showed no significant effects on group II mGluRs and NMDA. Further study needs to re-valulate effects of pure NAAG but it definitely looks like it has some pro mGluR3 actions.
NAAG is inactivated by a glial enzyme GCP-II, so when GCP-II is inhibited, mGluR3 signaling is increased through GCP-II -> NAAG -> mGluR3. GCP-II inhibition looks like one of the most promising ways of increasing mGluR3 firing, and inhibiting it has other benefits too. It looks more beneficial over directly administered NAAG, because administered NAAG seems to increase unpreferred firing.
GCP-II inhibition as well as a heterotropic mGluR/3 agonist restores short-term object recognition in mice treated with intoxicating doses (2.3 g/l) of ethanol. [Source]
Higher NAAG levels are associated with better cognition, suggesting that increasing NAAG levels through FOLH1/GCPII inhibition may improve cognition, and this has been shown consistently across primate and rodent models.
NAAG function is also highly correlated with IQ. For people with the gene rs202676, they have Decreased NAAG Levels (due to more GCP-II/FOLH1), and they have lower IQ on average as a result, likely due to lowered mGluR3 functioning [Source].
Inhibiting GCP-II also has been shown to be analgesic in many models (potentially with lasting effects), which makes sense as the dlPFC overlooks pain signaling, and GCP-II inhibition enhances dlPFC function. It has also been shown to decrease excitatory neurotransmission and inflammation-induced plasticity within the amygdala in a mouse model [Source]. It currently seems like GCP-II inhibition decreases glutamate release in the amygdala, however this has yet to be confirmed with microdialysis studies.
The best candidates for GCP-II inhibition look to be 2-PMPA and ZJ43. Both of them have proven useful in increasing delay cell firing, with 2-PMPA being able to double delay cell firing. 2-MPPA was a previous candidate, but it was withdrawn from use due to safety concern.
Both 2-PMPA and ZJ43 have been shown to significantly increase working memory in multiple animal models [Source] -tbf-.
2-PMPA looks the most potent GCP-II inhibitor (by a small margin), however it has bad BBB penetration and offtarget concerns at cytosolic carboxypeptidases (CCPs), significantly inhibiting Nna1 especially. This is structure specific to 2-PMPA so it is not GCP-II inhibition related.
Due to this, ZJ43 looks to be the most promising candidate currently for GCP-II inhibition and increasing mGluR3 firing. It has great predictions (should be taken with a grain of salt), and it has been proven effective in multiple studies. It also has better BBB penetration than 2-PMPA. Intranasal administration may also be the most effective method of delivery. The only downside to ZJ43 is that the doseage needed is quite high, possibly 75-100mg intranasally or 100-150mg orally.
Concerns Around Seretonin
Another potential concern to do with GCP-II inhibition is the effects on seretonin. In one rodent study, 2-PMPA was shown to reduce extracellular 5-HT (seretonin) in the dorsal striatum after multiple days of administration. However, there are many questions and things to be acknowledged here. First of all, this is a rodent study, and the dlPFC is much less developed in rodents. The dlPFC mediates emotional control, and increasing mGluR3 activity increases Delay Cell Firing, and increasing Delay Cell Firing has been shown to be beneficial for top-down control, as demonstrated with Guanfacine. Therefore, it needs to be considered that the rodents have a different effect profile than humans when mGluR3 is modulated.
Also another thing to consider is that any negative behavioral effects have yet to been replicated in human examples of GCP-II inhibition. This may be due to the lack of repeated-dose studies, a general lack of evidence, or a difference in human response to GCP-II inhibitors. 2-MPPA (later discontinued for chemical structure-related concerns) was well tolerated in a human trial only showing negative mood deficits after very high dosage (1125mg). However, it should be mentioned that this trial was not over multiple days.
Still, if seretonin is a potential concern, it may be considered that it is best to co-administer with something else that increases seretonin, such as Pinealon or similar. This concern may also be ameriolated with a mGluR2 NAM/Inhibitor as it would likely have positive effects on seretonin/5-HT2A.
mGluR2 Modulation
Even though mGluR3 can reduce glutamate via both presynaptic actions and glial action leading to consequent upregulated EAATs and glutamate uptake, but mGluR2 is much more potent at reducing glutamate transmission as compared to mGluR3. Excess glutamate associated excitotoxicity is implicated in Alzheimer's disease yet mGluR2 activation worsens amyloid-beta toxicity.
mGluR2 inhibition is also thought to be one mechanism of psychedelics that leads to antidepressant effects. In terms of cognition, mGluR2 but not mGluR3 has inverted dose-effect response on dlPFC Delay cell firing and WM improvements. Despite this, no mGluR2 NAM have been shown to have cognitive impairing effects. Thus, mGluR2 inhibition along with mGluR3 activation should fine tune our glutamate system.
mGluR2 inhibition seems to be the, or one of the downstream cognitively beneficial effect of MR (Mineralocorticoid Receptors) agonists [Source]. MR agonists are not viable due to side effects, however, it shows the potential for mGluR2's role to play in cognition. Studies using MR agonists have shown increased cognition [Source] accross a wide array of cognitive factors.
mGluR2 inhibition might also have a positive impact on sleep by increasing the threshold from wake to sleeping [ Source needed, I lost it ].
Neuroprotection
mGluR3-Based Protection
In several age-based decline models, improved motor activity and enhanced neuroprotection have been reported with mGluR3 agonists and GCP-II inhibitors.
mGluR3 Positive Allosteric Modulation can induce an increased production of GDNF and TGF-b [Source], in a mGluR3-dependent manner, which would also likely result after GCP-II inhibition as well.
GCP-II inhibition was shown to protect against hypoxia (up to 100%) in one study, and also protecting to a lesser extent against NMDA and glutamate injury. It was also shown to reverse cognitive memory deficits in one rodent study of Alzheimer’s disease [Source]. This makes sense as NAAG seems to be much lower in confirmed human Alzheimer’s cases, leading to lowered mGluR3 signaling.
GCP-II inhibition also reduced ischemia-induced nerve cell death in a cerebral occlusion stroke model and in a primary cell culture [Slusher et al., 1999][Tortella et al., 2000], and lowering GCP-II has been found to reduce ischemia-induced apoptosis [Source].
On top of that, GCP-II inhibition protects against ketamine-induced neurotoxicity at lower doses [Source], likely protective against schizophrenia and symptoms [Source] and more.
GCP-II inhibition has been shown to have a partial preventive effect on hyperalgesia and has also seems to be preventative against long-term diabetic neuropathy (in a rodent model) [Source]. ZJ-43 (GCP-II inhibitor) was shown to reduce neuronal degeneration in the CA3 region of the hippocampus 24 h after TBI (Traumatic Brain Injury), and reduced astrocyte death in the hippocampus [Source]
GCP-II inhibition has also been shown to increase lifespan by 15% in one study on mice [Source].